
近年来,国产创新药研发实力逐步提升,国内审评审批政策亦在优化。据国家药监局数据,2025年国内共批准上市76款创新药,相较2024年的48款增长58.3%,创下历史新高。
这一背景下,2026年全国两会期间,全国政协委员、复旦大学上海医学院副院长朱同玉建议:建立非预期药械严重不良事件风险共济制度。一方面由独立、专业的第三方机构监督落实不良反应报告制度,并对非预期性不良反应做出科学的评价、认定。另一方面建立保障制度,对这些被认定的非预期性严重不良反应提供补偿。
药品不良反应是指合格药品在正常用法用量下出现的,与用药目的无关的有害反应,是一种药品的固有属性。一般来说,所有药品都会存在或多或少、或轻或重的不良反应。所谓非预期的不良反应事件,可以简单理解为未知的、未载于临床试验药物研究者手册、上市后药品说明书中的不良反应。
朱同玉在接受界面新闻采访时介绍,当下,国内缺乏覆盖药品(含疫苗)及创新医疗器械产品全生命周期的严重不良事件补偿和风险共济制度。尤其在获批上市后的创新药械上,合法合规的补偿救济渠道不畅,被伤害人普遍补偿不足。这制约了创新药械的研发、注册审批及上市后的应用推广,也不利于创新医疗技术临床应用中的医患关系。
他向界面新闻提到,尤其是当下国内创新药发展迅猛,附条件获批的新药不在少数。这些药品上市后,药企仍需展开新的或继续正在进行中的临床研究,积累更多临床获益证据,从而转为常规批准。此时建立非预期药械严重不良事件风险共济制度恰有必要性。
界面新闻注意到,药品附条件批准是一种特殊审评方式,指的是具有突出治疗价值、临床急需的药物在未完成完整临床试验时,以“先批准后验证”的形式加快批准上市。换而言之,这实际上是在更多患者能尽快使用到新药,与新药安全性、疗效的充分验证之间做出平衡。
上世纪90年代起,药品附条件上市审批在美国、欧盟、日本等多国先后实施,成为一种国际上通行的审批方式,国内亦在2017年开始批准药品附条件上市申请。据丁香园insight数据库,2021年至2025年,每年均有20多款,共计超120款药物通过该方式获批。
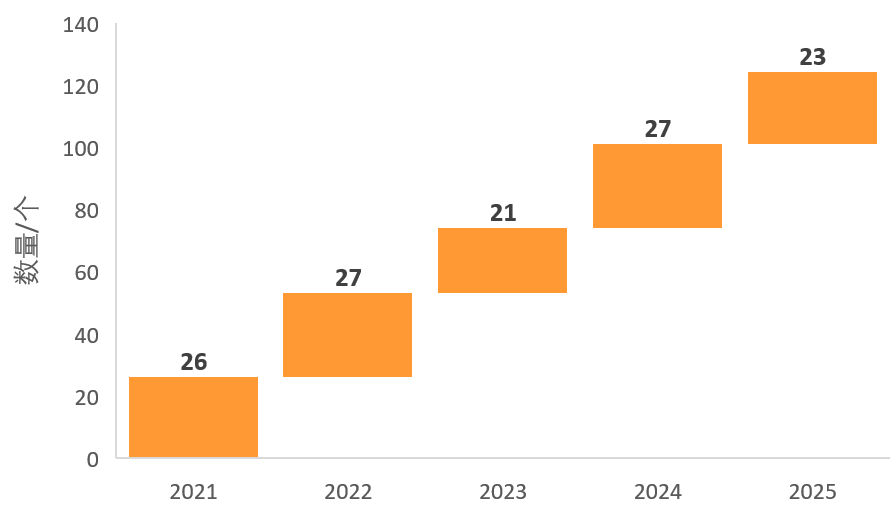
国内历年附条件批准品种数。数据来源:丁香园insight数据库
“比如干细胞、细胞疗法、噬菌体疗法这些新疗法现在看来没有什么问题(获益大于风险),但在大量群体、长期随访观察下,会不会出现(更多)风险还不知道。”朱同玉说。
他还向界面新闻举例,比如一款新药患者在使用后,在马路上遭遇车祸。这一个案中看不出新药与车祸之间的关联。但如果大量人群用药后都发生车祸,看似不相关的意外可能就与药物有关。比如药物是否具有致幻、致盲作用,造成使用者视野缺失、辨认不了路上的交通情况。此时即需要由独立第三方机构对药物与不良反应之间的相关性、因果关系做出科学认定,而非由一家药企、一家监管部门说了算。
实际上,这在药物开发和监管历史上并非天方夜谭,海外的“反应停”事件即是一个典型案例。上世纪50年代末,西德制药商梅瑞尔公司研制出一种镇静药物沙利度胺(即"反应停"),在欧洲、南美等地上市,用于缓解孕妇呕吐。随后,当地陆续出现“海豹肢”畸形婴儿。
直到1961年,德国医生通过流行病学研究证实这由反应停导致,该药随后退市,并在一定程度上推动完善了当时的药品监管政策。
亦有基因治疗领域人士告诉界面新闻,此前业内认为基因治疗在理论上可以实现“一次性治愈”,这点能吸引到很多患者。但实际并非如此,病人首先担心的是安全性问题。他以血友病为例,尤其目前已经有凝血八因子、九因子等可及的蛋白替代疗法,患者对新疗法的安全性有更多期待。
由此,朱同玉本次建议,在科学认定基础上,国家为这些非预期的严重不良反应建立风险共济的保障制度,例如可以考虑在国家层面设立专项公共基金池、企业缴纳一定的非预期严重不良反应保障基金、商业保险承保等形式。他提到,这一方面切实关系到公众的切身利益、生命健康,一方面也有利于创新药械公司大胆创新。
